Comprehensive Guide to the Three Types of Neurofibromatosis and Their Distinct Features
This comprehensive article explores the three main types of neurofibromatosis—NF1, NF2, and schwannomatosis—detailing their symptoms, genetic basis, diagnosis, and management strategies. Ideal for patients, healthcare providers, and researchers, it offers insights into these complex disorders and emphasizes the importance of early detection and personalized care for improved quality of life.

Neurofibromatosis (NF) is a complex genetic disorder characterized by the development of benign and sometimes problematic tumors along the nervous system, including the brain, spinal cord, and peripheral nerves. This condition affects individuals differently depending on its specific type, with each form presenting unique symptoms, risks, and challenges. Understanding the nuances of these three variants of neurofibromatosis is crucial for early diagnosis, management, and improving quality of life for those affected.
Neurofibromatosis manifests primarily in three distinct forms: NF1, NF2, and schwannomatosis (sometimes referred to as NF3). Each type has a different pattern of tumor growth, typical age of onset, and associated health issues, making accurate diagnosis essential for tailored treatment strategies.
Neurofibromatosis Type 1: The Most Common Form
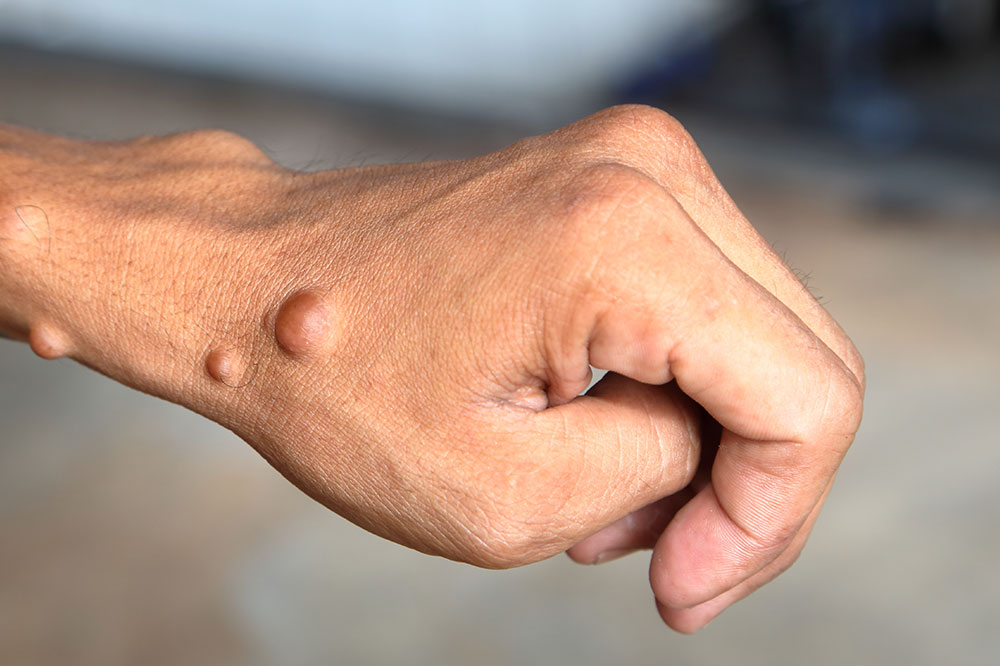
Neurofibromatosis Type 1 (NF1) is by far the most prevalent form, accounting for approximately 90% of all neurofibromatosis cases. It typically becomes apparent during childhood, although it can sometimes be diagnosed earlier or later in life. The hallmark characteristics of NF1 include distinctive skin features, specific tumor types, and potential skeletal abnormalities.
One of the primary signs of NF1 is the presence of multiple café-au-lait spots—flat, brown skin patches resembling coffee with milk—that often appear during early childhood. The number, size, and location of these patches can vary widely among individuals. Another characteristic feature are neurofibromas, which are soft, benign tumors that develop on or under the skin. These neurofibromas can be cutaneous (on the skin surface), subcutaneous (beneath the skin), or plexiform (more extensive, involving multiple nerve branches).
In addition to skin features, individuals with NF1 often develop Lisch nodules—small, benign tumors on the iris of the eye—which do not typically affect vision but are useful indicators for diagnosis. Bone deformities are also common, with scoliosis (curved spine) being a frequent complication. Some patients may experience learning disabilities or developmental delays, although intelligence is generally unaffected.
The genetic mutation responsible for NF1 affects the neurofibromin protein, which plays a critical role in regulating cell growth. This mutation leads to abnormal cell proliferation, resulting in tumor formation along the nervous system. The disorder is inherited in an autosomal dominant pattern, meaning a single copy of the mutated gene can cause the disease. However, new mutations can also occur spontaneously.
Neurofibromatosis Type 2: The Acoustic Nerve Tumor Syndrome
NF2 is less common than NF1 but presents with distinctive features primarily involving the auditory and vestibular nerves. Typically diagnosed in late adolescence or early adulthood, NF2 is characterized by the growth of benign tumors on the eighth cranial nerve, also known as the vestibulocochlear nerve. These tumors, called vestibular schwannomas or acoustic neuromas, impact hearing and balance.
Individuals with NF2 often experience progressive hearing loss that can start subtly and worsen over time. Tinnitus (ringing in the ears), dizziness, and balance problems are also common symptoms. As tumors grow, they can exert pressure on adjacent nerves and brain structures, potentially leading to facial weakness or numbness.
Early in the disease course, some individuals remain asymptomatic or have mild symptoms that are often overlooked. However, as the tumors enlarge, the effects become more pronounced, necessitating medical intervention. Cataracts are another feature frequently associated with NF2, often appearing earlier than age-related cataracts, and affecting vision.
The genetic mutation underlying NF2 affects the merlin (schwannomin) protein, essential for cellular growth control. Like NF1, NF2 is inherited in an autosomal dominant pattern, but spontaneous mutations are also observed. Regular screening, early detection, and surgical or radiotherapy options play pivotal roles in managing NF2.
Schwannomatosis (NF3): The Rare and Mysterious Form
Schwannomatosis, often classified as NF3, is a relatively rare form of neurofibromatosis that predominantly emerges during early adulthood. It is characterized by the development of multiple schwannomas—tumors arising from Schwann cells, which insulate nerve fibers—on cranial, spinal, and peripheral nerves. Unlike NF1 and NF2, schwannomatosis is less likely to involve tumors on the vestibular nerve, and hearing loss is less common.
The core symptom of schwannomatosis is chronic pain, which can be severe and localized depending on the tumor’s location. Patients may also report numbness, tingling, or weakness in affected areas, especially in fingers, toes, or limbs. Because symptoms are often subtle or attributed to other conditions, diagnosis can be delayed or challenging.
Histologically, schwannomas are benign, encapsulated tumors composed of Schwann cells. The mutations associated with schwannomatosis are different from those in NF1 and NF2, primarily involving mutations in the SMARCB1 and LZTR1 genes. It follows an autosomal dominant inheritance pattern but can also arise sporadically.
Due to the subtle and varied presentation, managing schwannomatosis involves regular monitoring via imaging studies, pain management, and, in some cases, surgical removal of tumors causing significant symptoms. Researchers are actively investigating targeted therapies to better control tumor growth and symptom progression.
Summary and Outlook
Neurofibromatosis remains a complex set of disorders with diverse manifestations that can impact multiple aspects of health and wellbeing. Early diagnosis through genetic testing and imaging can significantly improve management and outcomes. While there is no cure for neurofibromatosis, advancements in surgical techniques, targeted therapies, and supportive care have improved quality of life for many patients.
Ongoing research aims to understand the genetic mechanisms involved fully and develop innovative treatments that can halt or reverse tumor growth. Patients and families impacted by neurofibromatosis are encouraged to seek specialized medical care, genetic counseling, and support networks to navigate this multifaceted condition effectively.